













Executive summary: 10 Principles for Good AI Practice
Artificial intelligence is no longer discussed only as an emerging technology in drug development. It is increasingly becoming part of modern pharmaceutical development processes, including clinical trials, biomarker analysis, pharmacovigilance, manufacturing, and regulatory decision-making.
In January 2026, the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) jointly published ten guiding principles for Good AI Practice in medicine development, marking one of the first coordinated regulatory positions on artificial intelligence across the pharmaceutical industry. Learn more about AI regulation in clinical trials.
The publication signals a significant shift in how regulators approach AI. The discussion is no longer focused on whether AI can be used in medicine development, but on how it should be governed, validated, monitored, documented, and integrated into regulated environments.
For sponsors, CROs, and clinical trial partners, the principles for good AI practice provide an early framework for future expectations regarding:
- AI governance,
- data quality and traceability,
- lifecycle monitoring,
- human oversight,
- validation,
- and risk-based management.
This article explains the regulatory background behind the EMA/FDA initiative and explores what each of the ten principles for good AI practice may mean in reality for clinical trials and pharmaceutical development.
The regulatory background: how EMA’s AI framework evolved
The publication of the joint EMA/FDA principles did not appear suddenly. It is part of a broader European regulatory roadmap focused on data and artificial intelligence.
Earlier, the Network Data Steering Group (NDSG), operating within the European medicines regulatory network (EMRN), published a multi-year workplan for 2026–2028 focused on data and AI. The roadmap outlined the development of future guidance, terminology standardization, and governance frameworks for AI within the pharmaceutical sector.
The roadmap includes several key milestones presented below.
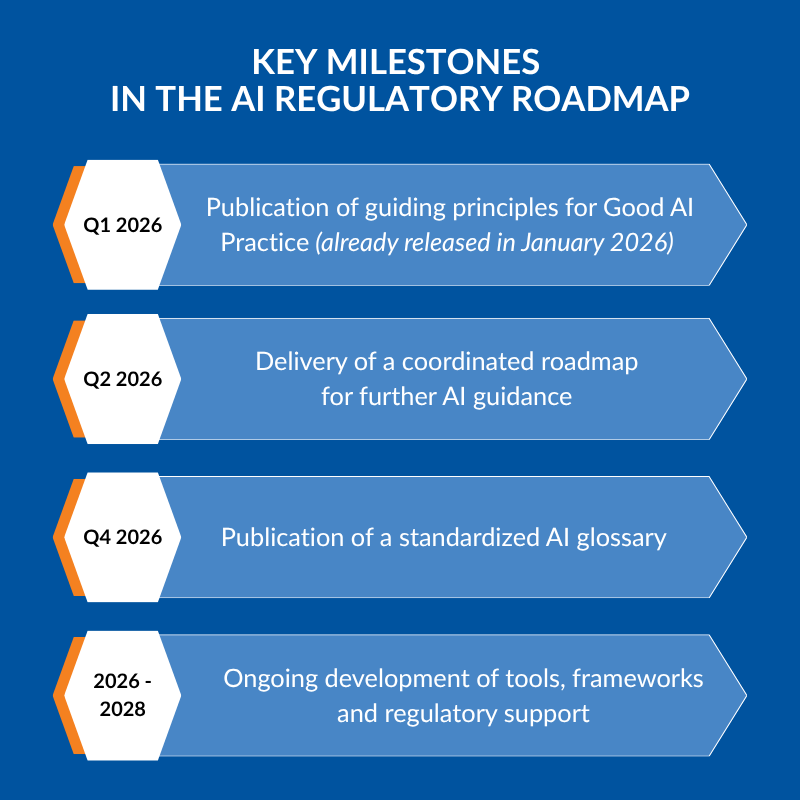
At the same time, EMA continued building on its earlier Reflection Paper on the use of AI in the medicinal product lifecycle, moving from high-level discussion toward practical governance expectations.
This progression shows that regulators increasingly treat AI as part of regulated pharmaceutical infrastructure rather than an experimental innovation area.
The 10 EMA/FDA Principles for Good AI Practice
As part of broader initiative, EMA and the U.S. Food and Drug Administration (FDA) jointly published ten principles for Good AI Practice in medicine development in January 2026. The document establishes a shared regulatory direction for how AI technologies should be developed, validated, monitored, and governed across the medicines lifecycle, including clinical trials, manufacturing, and pharmacovigilance.
According to the regulators, the principles are intended to support future international collaboration, harmonization efforts, and development of additional AI-related guidance and standards for the pharmaceutical industry.
1. “Human-centric by design”
“The development and use of AI technologies align with ethical and human-centric values.”
The first principle emphasizes that AI should support people rather than replace human responsibility and oversight. In clinical research, this means AI systems should be developed with patient safety, ethical considerations, and scientific integrity at the center of the process.
For sponsors and CROs, human-centric AI may include:
- maintaining expert review of AI-generated outputs,
- ensuring clinical relevance of AI-supported decisions,
- and reducing the risk of overreliance on automation.
The principle also reflects growing regulatory expectations regarding accountability in highly automated environments.
2. “Risk-based approach”
“The development and use of AI technologies follow a risk-based approach with proportionate validation, risk mitigation, and oversight based on the context of use and determined model risk.”
EMA and FDA clearly indicate that not all AI systems carry the same level of regulatory or operational risk. For example:
- AI used for workflow optimization may require lighter oversight,
- while AI supporting patient selection, endpoint interpretation, or safety analysis may require significantly stronger controls and validation.
For CROs and pharmaceutical companies, this principle strongly aligns AI governance with existing GxP and quality management approaches.
3. “Adherence to standards”
“AI technologies adhere to relevant legal, ethical, technical, scientific, cybersecurity, and regulatory standards, including Good Practices (GxP).”
This principle confirms that AI systems are expected to operate within existing regulated frameworks rather than outside them. For clinical trials, this may include alignment with:
- GCP,
- GCLP,
- computerized system validation practices,
- cybersecurity requirements,
- and data integrity expectations.
The message from regulators is clear: AI does not reduce compliance expectations. In many cases, it may increase the importance of documentation and governance.
4. “Clear context of use”
“AI technologies have a well-defined context of use (role and scope for why it is being used).”
Organizations using AI should clearly define:
- what the technology is intended to do,
- where it will be used,
- what data it depends on,
- and what limitations exist.
This becomes particularly important in clinical trials involving: decentralized workflows, biomarker analysis, patient stratification, or AI-supported operational decisions. A clearly defined context of use helps reduce misuse of models outside validated environments.
5. “Multidisciplinary expertise”
“Multidisciplinary expertise covering both the AI technology and its context of use are integrated throughout the technology’s life cycle.”
EMA and FDA emphasize that AI governance cannot be managed only by data scientists or IT teams.
Effective oversight requires collaboration between:
- clinical experts,
- regulatory specialists,
- quality assurance teams,
- statisticians,
- laboratory experts,
- software engineers,
- and operational teams.
For CROs and sponsors, this may increase the importance of cross-functional governance models during AI implementation.
6. “Data governance and documentation”
“Data source provenance, processing steps, and analytical decisions are documented in a detailed, traceable, and verifiable manner, in line with GxP requirements. Appropriate governance, including privacy and protection for sensitive data, is maintained throughout the technology’s life cycle.”
This principle is highly relevant for clinical research environments where data quality and traceability are critical. Regulators increasingly expect organizations to demonstrate:
- where data originated,
- how it was processed,
- what transformations were applied,
- and how AI-related decisions were made.
For central laboratories and global clinical trials, this reinforces the importance of: harmonized datasets, standardized workflows, audit readiness, and strong governance frameworks. The principle also highlights privacy and protection requirements for sensitive data.
7. “Model design and development practices”
“The development of AI technologies follows best practices in model and system design and software engineering and leverages data that is fit-for-use, considering interpretability, explainability, and predictive performance. Good model and system development promotes transparency, reliability, generalisability, and robustness for AI technologies contributing to patient safety.”
This principle focuses on the technical quality of AI systems. EMA and FDA expect organizations to follow structured development approaches supporting:
- reliability,
- transparency,
- robustness,
- reproducibility,
- and patient safety.
For pharmaceutical companies and CROs, this may require stronger integration between: software engineering, validation processes, and regulated operational environments. The mention of explainability and interpretability is particularly important for AI systems used in decision-support contexts.
8. “Risk-based performance assessment”
“Risk-based performance assessments evaluate the complete system including human-AI interactions, using fit-for-use data and metrics appropriate for the intended context of use, supported by validation of predictive performance through appropriately designed testing and evaluation methods.”
The regulators stress that AI performance assessment should evaluate not only the model itself, but the entire operational environment in which it functions. This includes:
- human interaction with the system,
- real-world workflows,
- testing methodologies,
- and context-specific performance metrics.
For clinical trials, this may become especially important for: imaging analysis, predictive biomarkers, patient recruitment algorithms and safety signal detection. The principle reinforces that validation should reflect actual operational use rather than ideal laboratory conditions.
9. “Life cycle management”
“Risk-based quality management systems are implemented throughout the AI technologies’ life cycles, including to support capturing, assessing, and addressing issues. The AI technologies undergo scheduled monitoring and periodic re-evaluation to ensure adequate performance (e.g., to address data drift).”
EMA and FDA clearly indicate that AI governance continues after deployment.
Organizations are expected to:
- monitor system performance,
- detect model drift,
- assess emerging risks,
- and periodically re-evaluate models.
For long-term clinical programs and global studies, lifecycle management may become essential to maintaining reliability and regulatory trust over time. This principle strongly aligns AI oversight with traditional pharmaceutical quality management systems.
10. “Clear, essential information”
“Plain language is used to present clear, accessible, and contextually relevant information to the intended audience, including users and patients, regarding the AI technology’s context of use, performance, limitations, underlying data, updates, and interpretability or explainability.”
The final principle focuses on communication and transparency.
Organizations should provide understandable information regarding:
- the AI system’s intended use,
- limitations,
- performance,
- underlying data,
- updates,
- and explainability.
Importantly, regulators emphasize that communication should be adapted to the intended audience, including users and patients. For sponsors and CROs, this may influence: documentation practices, training materials, vendor communication, regulatory submissions and audit preparation.
What these principles mean for clinical trials
The joint EMA/FDA publication suggests that regulators are preparing for broader AI integration across clinical development. Importantly, the document does not attempt to restrict innovation. Instead, it aims to ensure that AI systems operate within controlled, auditable, and scientifically reliable environments.
For clinical trials, the practical implications may include:
- greater focus on AI documentation,
- stronger expectations for data quality and traceability,
- integration of AI governance into quality systems,
- increased validation requirements,
- and closer collaboration between operational, technical, and regulatory teams.
The principles also reinforce the growing importance of: standardized data environments, harmonized laboratory processes, and robust lifecycle management. These areas are already highly relevant in modern multi-country studies and may become even more critical as AI-supported workflows continue expanding.
Conclusion
The publication of the joint EMA/FDA AI principles represents an important regulatory milestone for the pharmaceutical industry. For the first time, regulators on both sides of the Atlantic presented a coordinated vision of how AI should function within medicine development.
The key message is clear: AI is no longer treated as an experimental technology operating outside regulatory frameworks.
Instead, regulators increasingly expect AI systems to function within structured governance models focused on:
- transparency,
- oversight,
- validation,
- traceability,
- risk management,
- and accountability.
For sponsors, CROs, and clinical trial partners, this signals the beginning of a new phase where successful AI adoption will depend not only on technological capability, but also on operational readiness and regulatory alignment.
Learn more about AI in clinical trials:
FAQ – Principles for Good AI Practice
1. What are the “Principles for Good AI Practice” published by EMA and FDA?
The “Principles for Good AI Practice” are a joint set of ten guiding principles published by the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) in January 2026.
The document outlines how artificial intelligence technologies should be designed, validated, monitored, and governed throughout the medicines lifecycle, including clinical trials, manufacturing, and pharmacovigilance.
The principles focus on areas such as:
– risk-based management,
– data governance,
– lifecycle monitoring,
– transparency,
– human oversight,
– and regulatory compliance.
2. Why are the Principles for Good AI Practice important for clinical trials?
The Principles for Good AI Practice are important because they signal how regulators expect AI systems to operate within regulated clinical environments.
For sponsors and CROs, the principles provide early expectations regarding:
– AI validation,
– documentation,
– traceability,
– governance,
– and oversight.
They also indicate that AI in clinical trials will increasingly be evaluated within existing GxP and quality management frameworks.
3. Do the Principles for Good AI Practice apply only to clinical trials?
No. The Principles for Good AI Practice apply across the entire medicines lifecycle.
According to EMA and FDA, this includes:
– nonclinical research,
– clinical development,
– manufacturing,
– pharmacovigilance,
– regulatory decision-making,
– and post-marketing activities.
However, many of the principles are highly relevant for clinical trials because of the increasing use of AI in patient recruitment, biomarker analysis, imaging, operational forecasting, and safety monitoring.
4. Do the Principles for Good AI Practice introduce legally binding requirements?
At this stage, the Principles for Good AI Practice are not legally binding regulations.
Instead, they establish a common regulatory direction and foundation for future:
– guidance documents,
– standards,
– harmonization initiatives,
– and AI governance frameworks.
The principles are expected to influence future regulatory expectations and operational best practices across the pharmaceutical industry.
5. How do the Principles for Good AI Practice affect CROs and sponsors?
For CROs and pharmaceutical sponsors, the Principles for Good AI Practice may increase the importance of:
– AI governance structures,
– documentation processes,
– lifecycle monitoring,
– validation activities,
– and multidisciplinary oversight.
Organizations using AI technologies may need stronger collaboration between:
– operational teams,
– quality assurance,
– regulatory experts,
– laboratory specialists,
– and software or data science teams.
The principles also reinforce the importance of maintaining audit-ready and traceable AI-supported workflows.
6. What is the main regulatory message behind the Principles for Good AI Practice?
The main message is that regulators no longer treat artificial intelligence as an experimental technology operating outside regulated pharmaceutical environments.
Instead, the Principles for Good AI Practice show that EMA and FDA increasingly expect AI systems to function within structured governance models focused on:
– transparency,
– accountability,
– validation,
– traceability,
– and risk management.
For the pharmaceutical industry, this represents a transition from discussing AI innovation to governing AI within regulated clinical and operational processes.
References
- EMA and FDA set common principles for AI in medicine development, EMA, access date: 15.05.2026
- Guiding principles of good AI practice in drug development, EMA, FDA, access date: 15.05.2026
- Guiding Principles of Good AI Practice in Drug Development, FDA, access date: 15.05.2026
- Artificial intelligence, EMA, access date: 15.05.2026